
Symmetry in Action: Computations Predict Optimal Ligands for Uranyl Extraction. The complexation of uranyl nitrate with various trialkylphosphates was studied using density functional theory. Symmetrical ligands formed more stable complexes, and the predicted stability constants closely matched experimental values.
Read More: Inorg. Chem. 2025, (online).
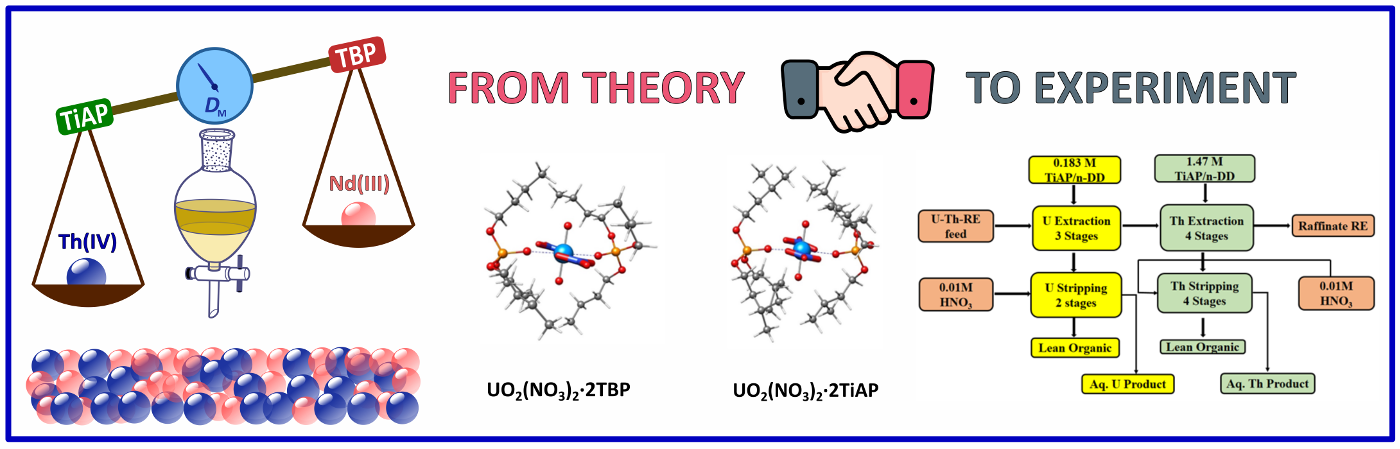
From Concept to Confirmation: The complexation behavior of trialkyl phosphates, TBP and TiAP with U(VI), Th(IV), and the representative rare earth ion Nd(III) were investigated at the density functional theory (DFT) level. The DFT calculations revealed a preferential extraction of U(VI) followed by Th(IV) over Nd(III), confirmed by extraction experiments. Geometric strain and dipole moment of the resulting complexes played a significant role in these findings.
Read More: J. Phys. Chem. A. 2024, 128(37), 7772-7784.
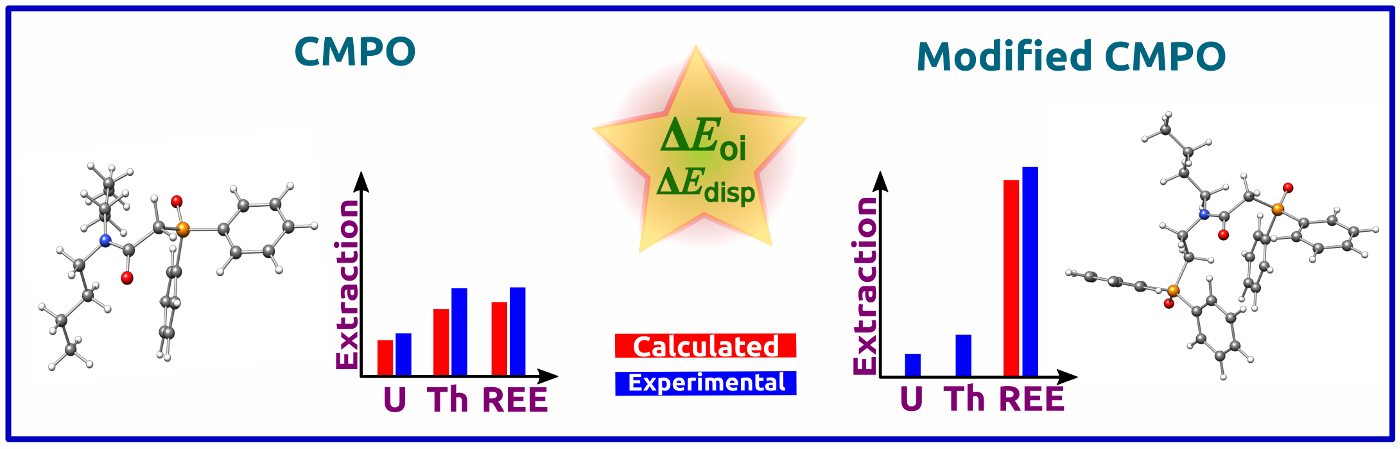
Complexation Behaviour of CMPO Ligands: The role of conventional and structurally modified carbamoylmethylphosphine oxide (CMPO) ligands towards the preferential extraction of Th(IV) over U(VI) and for REE(III) is investigated by applying density functional theory methodologies (DFT). The analysis centres around orbital and dispersion interactions and underscores the importance of these interactions in the design of the ligands.
Read More: J. Phys. Chem. A. 2024, 128(6), 1085–1097.
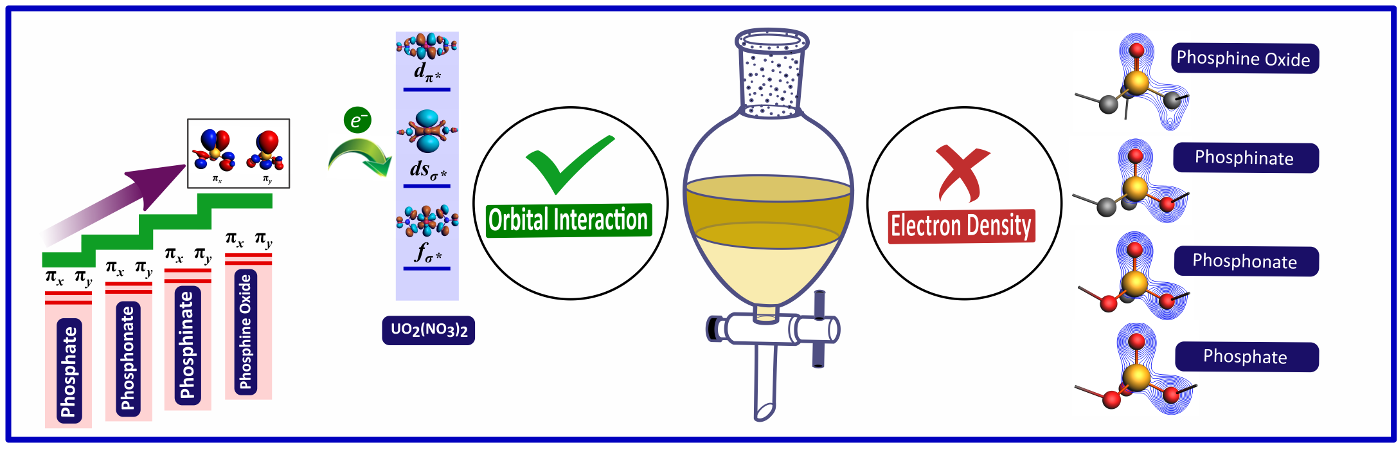
Orbital Interaction versus Electron Density of Phosphoryl Oxygen:
The organophosphorus compounds, namely trialkyl phosphates, phosphonates, phosphinates, and phosphine oxides, have been extensively used for complexing actinides. Hitherto, the assumption was that the enhanced electron density of phosphoryl oxygen (conventionally understood as the basicity of phosphoryl oxygen) in trialkyl phosphine oxides, when compared to phosphinates, phosphonates, and trialkyl phosphates, was primarily responsible for their better complexing behavior with actinides. However, for the first time, in the present study, we have demonstrated through quantum chemical calculations that "dispersion and orbital interactions" and not "phosphoryl oxygen electron density", are primarily responsible for the better complexing ability of phosphine oxides over other molecules.
Read More: J. Comput. Chem. 2023
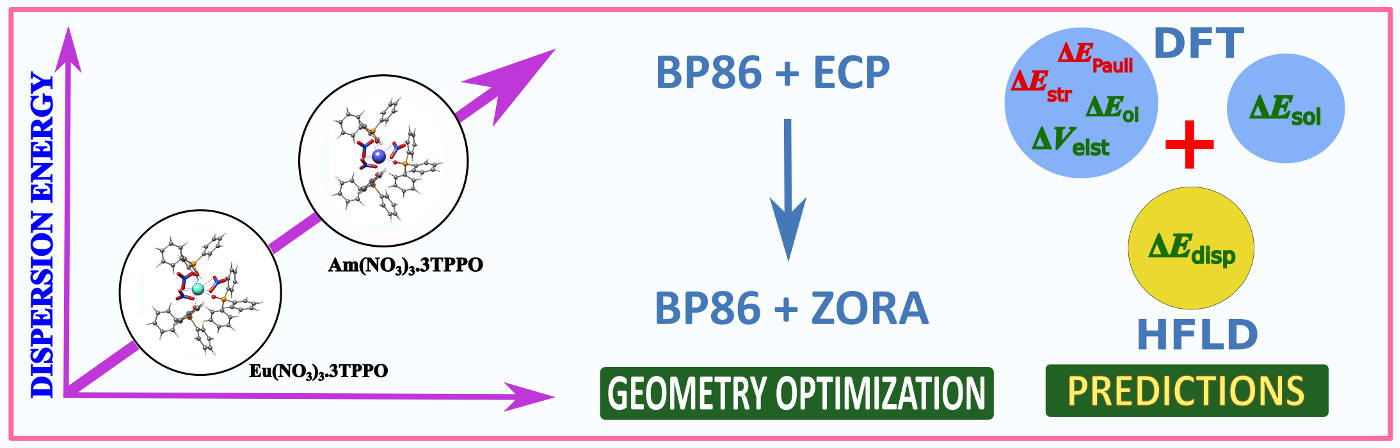
Dispersion Energy for Am(III) Complexes:The dispersion energy contribution for Am(III) complexes were evaluated for the first time by calculating the interfragment dispersion energies estimated at the CCSD level, by applying the London dispersion-corrected Hartree−Fock (HFLD) open-shell variant. The study highlights its importance in trivalent lanthanide & actinide complexes.
Read More: J. Phys. Chem. A. 2023
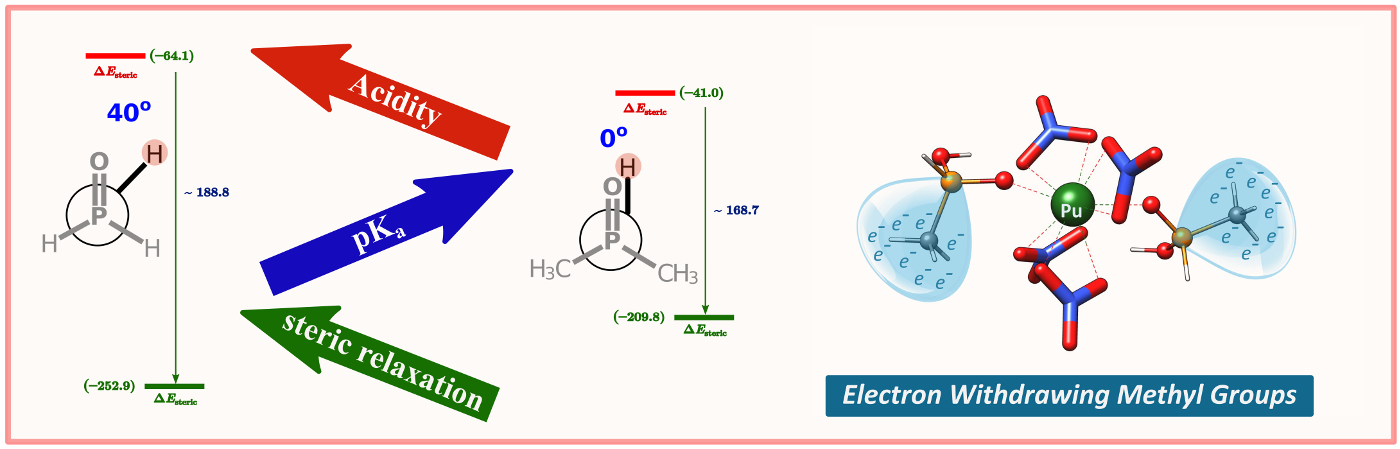
Electron Withdrawing Methyl Group: Quantum chemical calculations show that the methyl groups are electron-withdrawing in phosphinate ligands. The methyl-substituted phosphinic acids prefer Pu(IV) over U(VI), presumably a consequence of the increased covalent character of the metal-oxygen bond in Pu(IV) complexes.
Read More: Inorg. Chem. 2022
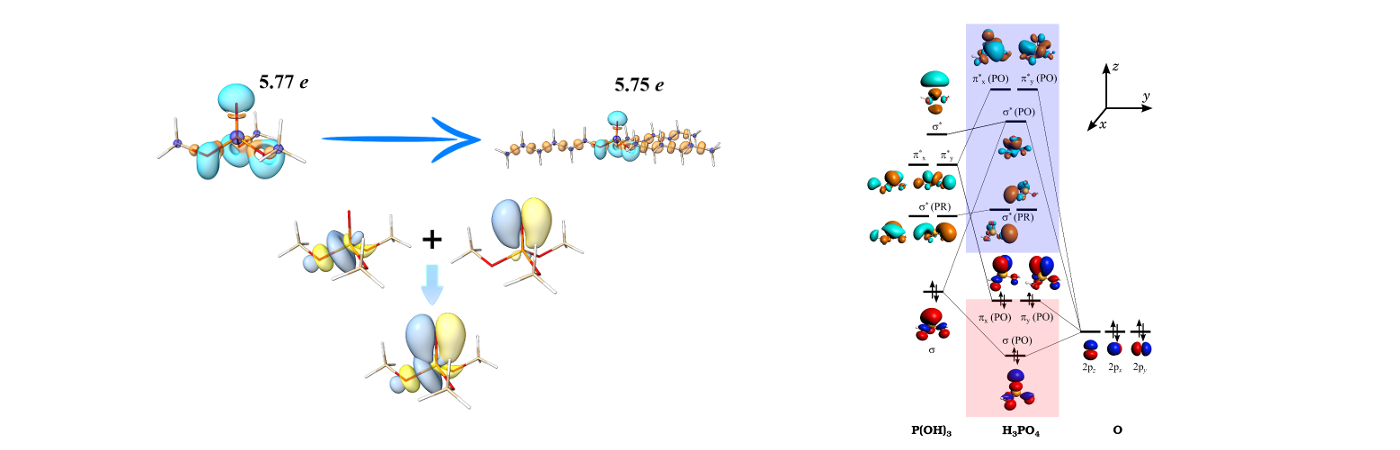
Phosphoryl Basicity vs Alkyl Chain Length: The effect of alkyl chain length on the basicity of phosphoryl oxygen in tri-alkyl phosphate ligands is investigated applying quantum chemical methodologies. The theoretical data suggests no convincing evidence for the correlation between the basicity of phosphoryl oxygen atom and alkyl chain length. Hyperconjugation interactions, which partially reduce π electron density of phosphoryl group, are estimated to be same for all ligands.
Read More: Chem. Phys. Lett., 2021.
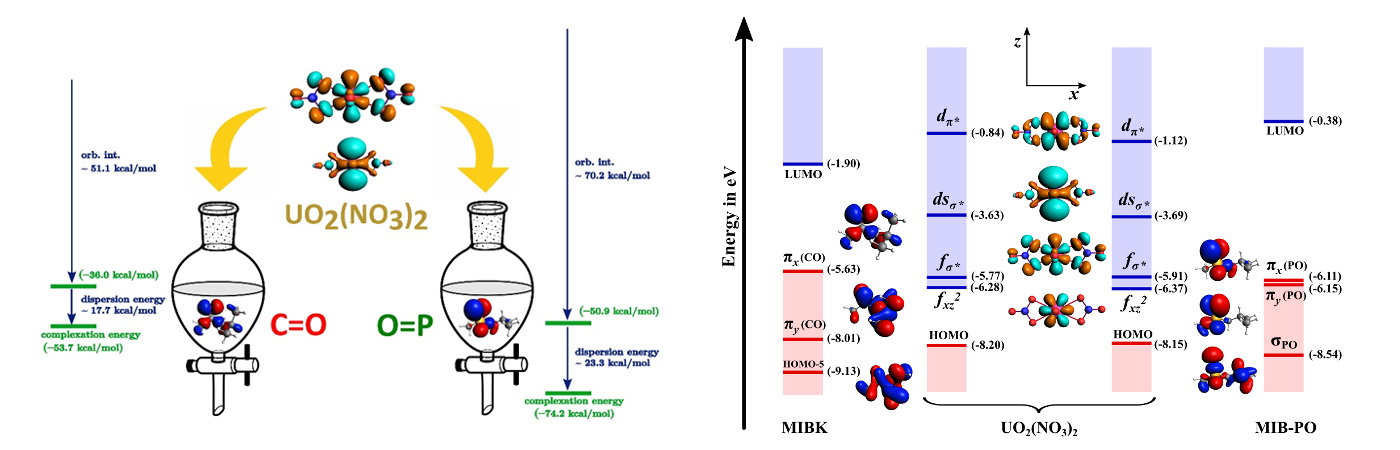
Carbonyl or Phosphoryl: Uranyl nitrate shows a preference for phosphoryl ligands over carbonyl. The quantum chemical calculations show that this is due to the enhanced stability of metal-phosphoryl bonding. The combined effect of orbital interactions and dispersion energy contributions is highlighted in this work.
Read More: J. Phys. Chem. A., 2020, 124(38), 7805-7815.